/reboot/media/9524b684-c4a9-11eb-9dc9-0242ac130004/952784cc-c4a9-11eb-8ba4-0242ac130004/0-0-pink-and-white-floral-textile-sy0wzqgmf8a-jpg.jpg)
Maladie de pompe
Maladie de pompe
Qu’est-ce que la Maladie de Pompe ?
Les maladies lysosomales (ML) constituent un groupe hétérogène de désordres métaboliques génétiques. L’insuffisance enzymatique généré entraîne une accumulation de son substrat dans le lysosome. Parmi ces pathologies, on distingue la glycogénose de type II dite maladie de Pompe.
Elle est due au déficit en alpha glucosidase ou maltase acide, entraînant une accumulation de glycogène dans le muscle squelettique et le myocarde. Il s'agit d'une maladie de surcharge lysosomale. Son spectre clinique est large allant de la forme pédiatrique à la forme adulte.
La forme pédiatrique (dite infantile) débute dans les premiers mois de la vie. Elle est caractérisée par une atteinte musculaire et cardiaque sévère. La forme adulte peut débuter à tout âge, habituellement sans atteinte du myocarde.
Il existe des patients symptomatiques ou asymptomatiques en fonction du degré d’activité de l’enzyme mutée GAA (alpha glucosidase acide) dans cette pathologie. La relation entre l’activité de l’enzyme et la gravité de la maladie est inversement proportionnelle.
Epidémiologie de cette glycogénose
La maladie de pompe est une maladie très rare. La prévalence de cette glycogénose est de 1/40 000.
Étiologie et physiopathologie de cette maladie rare
La maladie de Pompe est une maladie neuromusculaire d’origine génétique à transmission autosomique récessive.
Elle est due à une mutation du gène GAA codant pour l’alpha glucosidase acide localisée sur le chromosome 17.
Cette mutation conduit à une insuffisance en GAA, l’enzyme qui permet d’hydrolyser le glycogène lysosomal en glucose. Ainsi le manque de cette enzyme conduit à une accumulation intra-lysosomale de glycogène.
Au fil du temps, les lysosomes deviennent de plus en plus nombreux et de plus en plus volumineux. Est en cause l’accumulation de cette macro molécule de glycogène. Ce dernier va également s'accumuler hors des lysosomes, dans le cytoplasme des cellules. Celles-ci vont progressivement s'engorger, entraînant une altération du fonctionnement de différents organes, en particulier les muscles squelettiques.
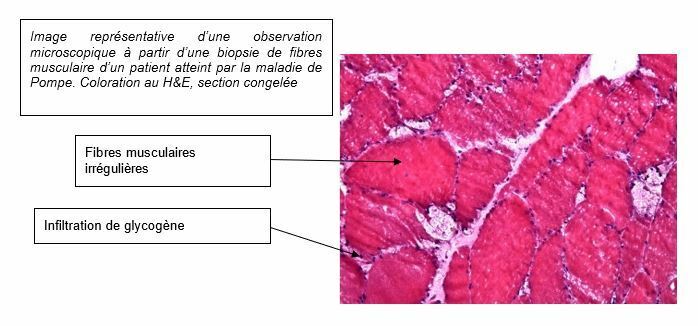
Sur une coupe microscopique, il est possible de mettre en évidence l’augmentation de la taille des vacuoles et la nécrose des myocytes suite à l’infiltration de glycogène
Dans la glycogénose de type II, il existe un risque supplémentaire de fragilisation des myocytes. En effet, la pression exercée par une contraction musculaire importante, comme un exercice physique intense, peut comprimer les lysosomes jusqu'à augmenter le déversement du glycogène lysosomal dans le cytoplasme du myocyte, entraînant la rupture de la cellule.
L'accumulation de glycogène touche également les muscles lisses de l’appareil urinaire, ainsi que ceux présents tout au long du tube digestif.
Elle peut aussi concerner les fibres nerveuses entériques contrôlant les muscles lisses digestifs ou les cellules de Schwann fabriquant la gaine de myéline isolante et protectrice de ces fibres nerveuses.
Le déficit est ubiquitaire mais il est exprimé principalement par certains organes : le cœur et/ou le muscle squelettique.
Certaines mutations (ex: c.-32-13T> G) sont majoritaires mais l’identification de nombreuses mutations est en accord avec l'hétérogénéité clinique.
Quelles sont les formes de la Maladie de Pompe ?
Protéiforme, elle peut être de forme infantile, adulte ou intermédiaire. Le tableau clinique est en fonction des sites affectés par la surcharge.
Forme Infantile
Début très précoce avant l’âge de 6 mois voire dès la période anténatale.
C’est la forme la plus sévère. Sans traitement spécifique, l’espérance de vie est très courte, inférieure à deux ans dans la plupart des cas. L’examen révèle une atteinte cardiaque et musculaire importante.
- Cardiomégalie: cardiomyopathie hypertrophique évoluant vers une insuffisance cardiaque,
- Insuffisance respiratoire par dysfonctionnement du diaphragme ou par compression d’une bronche due à la cardiomégalie,
- Hypotonie majeure,
- Infection respiratoire à répétition,
- Hypo mobilité,
- Difficulté de la succion et de la déglutition,
- Difficulté alimentaire,
- Retard de croissance,
- Absence de réflexes ostéotendineux,
- Signes de surcharge lysosomale : hépatomégalie et macroglossie.
Forme intermédiaire dite juvénile
Les signes cliniques apparaissent de manière plus tardive que dans la forme pédiatrique. L’âge des enfants au diagnostic est très variable allant de 1 an jusqu’à la forme adulte.
Elle progresse plus lentement. L’espérance de vie des enfants et adolescents est très variable. Il est difficile de donner une estimation. Toutefois, un enfant touché entre l’âge de 2 et 20 ans peut vivre longtemps si les troubles respiratoires sont pris en charge.
Les symptômes sont les suivants :
Les signes cliniques apparaissent plus tardivement que dans la forme infantile, l’âge au diagnostic est très variable allant de 1 an jusqu’à la forme adulte :
- Faiblesse musculaire ou myalgie prédominant au niveau des ceintures avec une élévation des enzymes musculaires : créatine phosphokinase (CPK),
- Réflexes ostéotendineux diminués ou abolis,
- Retard d'acquisition motrice sans atteinte cognitive,
- Troubles respiratoires : dyspnée, apnée du sommeil, infection récidivante ,
- Trouble de la déglutition.
Dans la forme juvénile, l’atteinte du coeur est quasiment inexistante et très modérée.
Forme Adulte
L’espérance de vie est très variable. Elle dépend de la sévérité de l’atteinte pulmonaire.
Le tableau clinique est le plus souvent caractérisé par une myopathie des ceintures avec atteinte précoce de la musculature respiratoire et de la musculature lisse induisant:
- Atteinte musculaire :
- Faiblesse proximale des ceintures prédominant à la ceinture pelvienne, se traduisant initialement par des difficultés à la marche, pour se relever d’un siège ou monter les escaliers.
- Faiblesse de la musculature axiale fréquente et précoce, se manifestant par une hyperlordose.
- Plus rarement : intolérance à l’effort et/ou myalgies.
- Atteinte respiratoire sous forme d’insuffisance respiratoire restrictive parfois aiguë en l’absence même de toute atteinte musculaire cliniquement patente,
- Élévation isolée du taux de CPK,
- Atteinte cardio-vasculaire : rare et peu sévère chez l’adulte : cardiomyopathie hypertrophique (hypertrophie ventriculaire gauche concentrique modérée, non obstructive), troubles conductifs le plus souvent modérés.
- Atteinte pharyngée et gastroentérologique.
Du fait de la facilité du dépistage et de l’existence d’un traitement spécifique, la maladie de Pompe doit être évoquée devant tout déficit des ceintures.
Comment diagnostiquer la maladie de Pompe ?
L'identification de la maladie de pompe est souvent tardive à cause de sa rareté et sa similarité avec d'autres maladies neuromusculaires. Ainsi le diagnostic de certitude repose sur le résultat d'un ensemble d’examens cliniques et biologiques :
Anamnèse
- Existence d’un historique familial,
- Mise en évidence de symptômes évocateurs comme l’existence d’une hypotonie ou d’une gêne respiratoire.
Examens pour un diagnostique de certitude
- Mesure de l’activité de l’alpha glucosidase acide sur cellules sanguines ou fibroblastes de biopsie cutanée.
- Test génétique par séquençage du gène GAG codant l'alpha-glucosidase acide, situé sur le chromosome 17 en position q25.2, et comportant 20 exons.
- Le tétraglucose (Glc4) urinaire est un biomarqueur non spécifique de la maladie de Pompe. Le résultat est élevé pour les patients atteints de la maladie de Pompe, quel que soit leur âge.
Un dosage est préconisé au moment du diagnostic.
Examens Complémentaires
- Dosage des enzymes musculaire : Dosage des enzymes musculaires : la créatine kinase est libérée lors de la lyse du myocyte, c’est le cas dans les myopathies, dans la maladie de Pompe, le taux de CPK présente une élévation modérée (< 5 fois la valeur normale),
- Electromyogramme: mise en évidence de l’origine myopathique de l’atteinte de la fibre musculaire,
- IRM : présence d’une infiltration graisseuse et d’une atrophie des muscles atteints,
- Biopsie musculaire: mise en évidence de l’accumulation de glycogène dans les lysosomes musculaires, dans un stade avancé on mets également en avant la présence de glycogène dans le cytoplasme des cellules musculaires.
Cet examen est réalisé lorsque l’origine des symptômes musculaires n'apparaît pas clairement.
- Électrocardiogramme: pouvant mettre en évidence, surtout dans la forme pédiatrique, un espace PR court, des complexes QRS hypervoltés, et des anomalies de la repolarisation avec risque d’arythmie supraventriculaire et ventriculaire,
- Bilan respiratoire : gaz du sang capillaire, une oxymétrie nocturne et/ou une capnométrie nocturne ou au mieux une polysomnographie.
Comment soigner la maladie de Pompe ?
Il existe des traitements pour soulager au mieux les manifestations de la glycogénose de type II.
En France, dix médicaments ont obtenu l’autorisation de mise sur le marché (AMM) dans sept maladies lysosomales différentes. Grâce à la recherche, de nombreux progrès ont permis la connaissance physiopathologique des maladies lysosomales induisant le développement de solutions thérapeutiques efficaces. Néanmoins, la plupart des maladies lysosomales restent encore orphelines, sans traitement spécifique. Les thérapeutiques palliatives restent indispensables pour ralentir ou stabiliser le cours des maladies ou en corriger les symptômes.
Dans la maladie de Pompe, l’approche thérapeutique dominante est l'enzymothérapie substitutive (EST). Elle permet de diminuer la surcharge et d'améliorer, voire de limiter la progression de l’atteinte viscérale et de ses conséquences cliniques sur la santé des patients.
L'enzymothérapie substitutive par l'alglucosidase alpha a fait l'objet de plusieurs essais ces dernières années sur le plan international. L'enzymothérapie montre une efficacité parfois spectaculaire dans les formes pédiatriques. Elle est donnée à un rythme régulier (1 fois chaque 7 jours ou chaque 14 jours) par voie intraveineuse sur une ou quelques heures en fonction de la molécule, de la dose à injecter et de la tolérance du patient. Le suivi de la maladie est réalisé par des médecins et centres spécifiques.
Le registre français de la maladie de pompe recense l'ensemble des cas en France atteint de la maladie de Pompe. Il recommande de collecter toutes les données des patients français recevant l'alglucosidase alpha.
Selon les résultats des deux études publiées en juin 2020 qui s'appuient notamment sur le registre, après 18 mois de traitement, l’efficacité de l’enzymothérapie substitutive est confortée. Ainsi, le traitement par enzymothérapie présente les résultats suivants pour les patients :
- améliore la fonction motrice au cours des 3 premières années de prise du traitement (avec une amélioration de la distance de marche au test de 6 minutes de marche) suivi d’un déclin progressif ;
- ralentit ou stabilise la progression de l’atteinte respiratoire sur un suivi de 5 ans.
Conclusion
La maladie de pompe est une pathologie très hétérogène de part ses signes cliniques, son retentissement sur la vie sociale. En effet, la maladie est protéiformes pouvant être très évoluée en mettant en péril le pronostic vital pour la forme pédiatrique ou asymptomatique pour des patients dont la découverte de la maladie est fortuite. Une approche multidisciplinaire est à mettre en place par le médecin traitant alliant d'autres professionnels de santé tels que kinésithérapeute, pneumologue, cardiologue et pharmacien.
Si vous êtes un professionnel de santé, inscrivez-vous pour découvrir des vidéos sur la Maladie de Pompe.
Tous droits de reproduction réservés.
Sources
- https://www.has-sante.fr/upload/docs/application/pdf/2016-08/pnds_-_maladie_de_pompe.pdf
- https://pubmed.ncbi.nlm.nih.gov/30155607/
- https://www.myobase.org/doc_num.php?explnum_id=17087
- https://pubmed.ncbi.nlm.nih.gov/16898258/
- Long-term benefit of enzyme replacement therapy with alglucosidase alfa in adults with Pompe disease: prospective analysis from the French Pompe Registry. Semplicini C, De Antonio M, Taouagh N, et al. J Inherit Metab Dis. 2020 (Juin)
- Respiratory function during enzyme replacement therapy in late-onset Pompe disease: longitudinal course, prognostic factors, and the impact of time from diagnosis to treatment start. Stockton DW, Kishnani P, van der Ploeg A, et al. J Neurol. 2020 (Juin).
:strip_exif()/reboot/media/9524b684-c4a9-11eb-9dc9-0242ac130004/617562d2-d65a-11ee-97ad-0242ac12000a/1-1-vous-souhaite-de-bonnes-vacances-15.png)
/reboot/media/9524b684-c4a9-11eb-9dc9-0242ac130004/82d45c8a-cfa8-11ed-a2f8-0242ac14000c/1-1-2aj9xm6.jpg)
/reboot/media/9524b684-c4a9-11eb-9dc9-0242ac130004/0f7b961e-f2dc-11ef-9a0a-3260d44c31e4/1-1-2rh22t4.jpg)